Density Matrix Renormalization Group
What is DMRG?
The Density Matrix Renormalization Group (DMRG) was introduced by Steven White in 1992 and quickly became the method of choice for one-dimensional quantum lattice models. DMRG finds the ground state variationally within the family of matrix product states (MPS): wave functions of the form
where each is a matrix of dimension at most . The integer — called the bond dimension — controls the accuracy of the approximation: is a product state, and increasing captures more quantum entanglement.
Why does this work so well in 1D? Gapped ground states of local Hamiltonians satisfy an area law: the entanglement entropy of any contiguous block of sites is bounded by a constant independent of block size. This means the exact ground state can be well approximated by an MPS with a fixed, moderate . For the Heisenberg chain (which is gapless), the entanglement entropy grows only logarithmically, so – is typically sufficient.
DMRG optimizes the matrices by sweeping: it starts from one end of the chain, optimizes the matrices at each site in turn while holding all others fixed, then sweeps back from the other end, repeating until convergence. Each optimization step is a small eigenvalue problem — far cheaper than the full diagonalization that ED requires. The cost of a DMRG sweep scales as , where is the local Hilbert space dimension, versus for ED. For and , ED would require billion states; DMRG with is entirely routine.
Why open boundary conditions? DMRG is far more efficient on chains with open boundaries (OBC) than with periodic boundary conditions (PBC). With PBC, the entanglement structure wraps around the chain, roughly doubling the effective bond dimension needed for the same accuracy. OBC is therefore the standard choice for DMRG calculations.
The physical model
We study the antiferromagnetic Heisenberg chain,
on sites with open boundary conditions. Unlike the chain (which has the Haldane gap), the chain is gapless: its low-energy spectrum forms a Tomonaga-Luttinger liquid (TLL) with power-law correlations. Its exact ground-state energy per bond in the thermodynamic limit is given by the Bethe ansatz:
This is one of the most-studied benchmark systems in computational quantum physics and an ideal starting point for learning DMRG.
Running the simulation
The entire setup, run, and analysis fits in a single Python script:
import pyalps
import numpy as np
import matplotlib.pyplot as plt
import pyalps.plot
parms = [ {
'LATTICE' : "open chain lattice", # open boundary conditions (better for DMRG)
'MODEL' : "spin",
'CONSERVED_QUANTUMNUMBERS' : 'N,Sz', # exploit N and Sz conservation to block-diagonalize
'Sz_total' : 0, # target the Sz=0 sector (ground state)
'J' : 1, # antiferromagnetic exchange
'SWEEPS' : 4, # number of DMRG sweeps (left→right→left = 1 sweep)
'NUMBER_EIGENVALUES' : 1, # find only the ground state
'L' : 32, # chain length
'MAXSTATES' : 100 # bond dimension m (key accuracy parameter)
} ]
input_file = pyalps.writeInputFiles('parm_spin_one_half', parms)
res = pyalps.runApplication('dmrg', input_file, writexml=True)Parameter notes
MAXSTATESis the most important accuracy parameter. It sets the maximum bond dimension . Increasing it improves accuracy at the cost of compute time (which scales as ). For this 32-site benchmark, gives a truncation error well below .SWEEPSis the number of full left-to-right-and-back sweeps. Four sweeps is enough to demonstrate convergence here; production calculations often use 10–20 sweeps, or sweep until the energy change per sweep falls below a threshold.CONSERVED_QUANTUMNUMBERS: 'N,Sz'tells DMRG to exploit both total particle number and total magnetization as good quantum numbers, block-diagonalizing the MPS matrices. This makes the calculation faster and more numerically stable.NUMBER_EIGENVALUES: 1targets only the ground state. Increasing this allows DMRG to target the lowest few eigenstates simultaneously, at additional cost.
Loading ground state observables
After the simulation, loadEigenstateMeasurements retrieves all observables measured by the DMRG code — energy, magnetization, correlation functions, and any other quantities ALPS computed for the final converged ground state:
data = pyalps.loadEigenstateMeasurements(pyalps.getResultFiles(prefix='parm_spin_one_half'))
for s in data[0]:
print(s.props['observable'], ' : ', s.y[0])Each element s in data[0] corresponds to one observable. s.props['observable'] is its name (e.g., 'Energy', 'Magnetization') and s.y[0] is its value. The ground-state energy should be close to for this 32-site open chain.
Loading iteration history
To understand how the algorithm converged, we load per-sweep measurements:
itr = pyalps.loadMeasurements(pyalps.getResultFiles(prefix='parm_spin_one_half'),
what=['Iteration Energy', 'Iteration Truncation Error'])itr[0][0] contains the energy recorded at each half-sweep; itr[0][1] contains the truncation error at each step. The truncation error is the sum of the discarded eigenvalues of the reduced density matrix — the weight in the exact ground state that the MPS with states cannot represent. A well-converged calculation should reach a truncation error below ; for on this system, values around – are typical.
Plotting convergence
plt.figure()
pyalps.plot.plot(itr[0][0])
plt.title('Iteration history of ground state energy (S=1/2)')
plt.ylim(-15, 0)
plt.ylabel('$E_0$')
plt.xlabel('Iteration')
plt.figure()
pyalps.plot.plot(itr[0][1])
plt.title('Iteration history of truncation error (S=1/2)')
plt.yscale('log')
plt.ylabel('Truncation error')
plt.xlabel('Iteration')
plt.show()Results
The energy converges rapidly to a plateau within the first few sweeps. The plot below shows the energy as a function of iteration number — it stabilizes well before the final sweep:
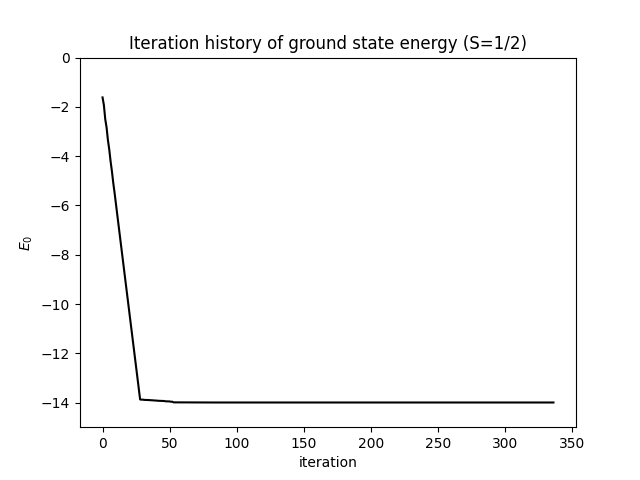
The truncation error decays monotonically on a logarithmic scale, reflecting the improving quality of the MPS approximation with each sweep:
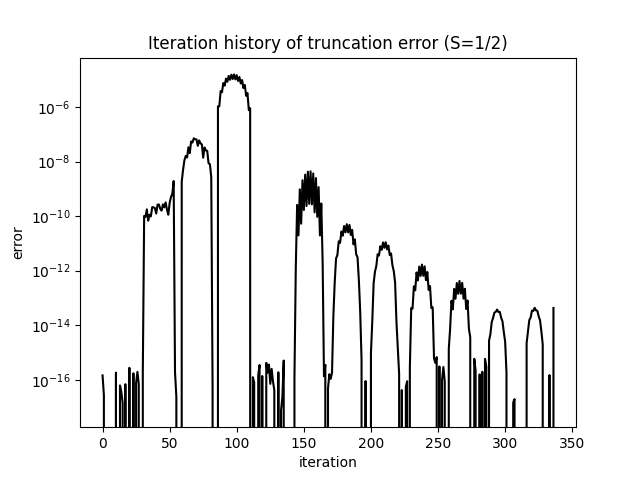
Once both curves have flattened, the calculation is converged. To improve accuracy further, increase MAXSTATES and verify that the energy and truncation error change negligibly.