Exact Diagonalization
What is exact diagonalization?
Exact diagonalization (ED) is the most direct approach to solving a quantum many-body problem: build the Hamiltonian matrix explicitly in a basis of many-body states, then find its eigenvalues and eigenvectors. The result is numerically exact — no approximations, no sign problem, no sampling error. The catch is cost: for a spin- chain of sites, the Hilbert space has dimension , which grows exponentially. For this means million states for , and billion for — already at the edge of what is tractable even on large computers.
The ALPS sparsediag code addresses this with sparse diagonalization via the Lanczos algorithm: instead of diagonalizing the full matrix, it builds a small Krylov subspace from repeated matrix-vector products and extracts the lowest few eigenvalues from it. This is practical because the Heisenberg Hamiltonian is extremely sparse — each basis state is connected to only others (one per bond), so each matrix-vector product costs rather than .
ED complements QMC: it gives exact ground-state and low-energy spectra at zero temperature, works for any model regardless of sign problems, but is limited to system sizes accessible by Lanczos ( for ).
The physical model: the S=1 Heisenberg chain
This tutorial studies the antiferromagnetic Heisenberg model on a one-dimensional chain,
where each is a spin-1 operator. This model describes a chain of magnetic moments with antiferromagnetic nearest-neighbor exchange.
The Haldane conjecture
In 1983, Duncan Haldane made a surprising prediction: the ground state of integer-spin antiferromagnetic chains () is gapped — there is a finite energy cost to create any excitation above the ground state. Half-integer chains (), by contrast, are gapless, with a continuous spectrum of low-energy excitations all the way down to zero energy.
This was unexpected because classical intuition and simple mean-field arguments suggest both cases should behave similarly. The distinction arises from a topological contribution in the quantum field theory description that is present for half-integer but absent for integer spin. Haldane’s conjecture was initially controversial, but it has since been confirmed numerically to high precision and experimentally in materials such as Ni(CHN)NO(ClO) (NENP).
For the chain, the gap converges to
in the thermodynamic limit (). On a finite chain of length , the apparent gap is larger and closes exponentially toward the thermodynamic value:
where is the correlation length of the Haldane phase. Because is not small, chains up to still show significant finite-size corrections — this is why a careful extrapolation is needed.
Strategy
We will:
- Run
sparsediagon chains of length . - For each , compute the lowest energy in the sector (singlet ground state) and the sector (lowest triplet state).
- Form the gap .
- Fit the exponential finite-size formula and extract .
Imports
import pyalps
import numpy as np
import matplotlib.pyplot as plt
import pyalps.plot
import pyalps.fit_wrapper as fwQuantum number sectors
Before looking at the code, it is worth understanding why we run two separate diagonalizations per chain length — one with Sz_total=0 and one with Sz_total=1.
The Heisenberg Hamiltonian commutes with the total magnetization , meaning total is conserved: . The full Hilbert space therefore block-diagonalizes into independent sectors labelled by . We can diagonalize each block independently.
This is the key to making ED tractable. For , , the full Hilbert space has dimension million; the sector has dimension roughly million — smaller, but crucially, ALPS also exploits translational symmetry and parity, reducing the effective dimension by another factor of . Working in sectors rather than the full space is essential.
The singlet ground state of the antiferromagnet lies in . The lowest triplet excited state — the first state with total spin 1 — appears as the ground state of the sector. Their energy difference is the triplet gap.
Parameters and input files
parms = []
for l in [4, 6, 8, 10, 12, 14, 16]:
for sz in [0, 1]:
parms.append(
{
'LATTICE' : "chain lattice",
'MODEL' : "spin",
'local_S' : 1, # S=1 spin chain
'J' : 1, # antiferromagnetic exchange (sets energy scale)
'L' : l, # chain length
'CONSERVED_QUANTUMNUMBERS' : 'Sz', # tell ALPS to block-diagonalize in Sz
'Sz_total' : sz # target sector (0=singlet, 1=triplet)
}
)
input_file = pyalps.writeInputFiles('parm2a', parms)writeInputFiles creates the XML input files in the ALPS format that sparsediag reads. The prefix 'parm2a' is used to name the output files.
Running the solver
res = pyalps.runApplication('sparsediag', input_file)sparsediag runs the Lanczos algorithm on each parameter set. For each combination of , it further block-diagonalizes within the sector using translational symmetry — so it actually solves one smaller matrix per lattice momentum . The output is stored in HDF5 files named after the prefix.
Loading results and understanding the data structure
data = pyalps.loadSpectra(pyalps.getResultFiles(prefix='parm2a'))data is a list of simulations, one per parameter set. Each element sim in data is itself a list of DataSet objects — one per lattice-momentum subsector within the chosen sector. Each DataSet carries:
sec.props: the parameters for this run (including'L','Sz_total','TOTAL_MOMENTUM').sec.y: a NumPy array of eigenvalues found in this -subsector.
Extracting the gap
The ground state in each sector can have any lattice momentum, so we collect all eigenvalues across all -subsectors and take the global minimum:
lengths = []
min_energies = {}
for sim in data:
l = int(sim[0].props['L'])
if l not in lengths: lengths.append(l)
sz = int(sim[0].props['Sz_total'])
all_energies = []
for sec in sim: # loop over k-subsectors within this (L, Sz) run
all_energies += list(sec.y)
min_energies[(l, sz)] = np.min(all_energies)After this loop, min_energies[(l, 0)] is the ground-state energy in the singlet sector and min_energies[(l, 1)] is the ground-state energy in the triplet sector, for each chain length .
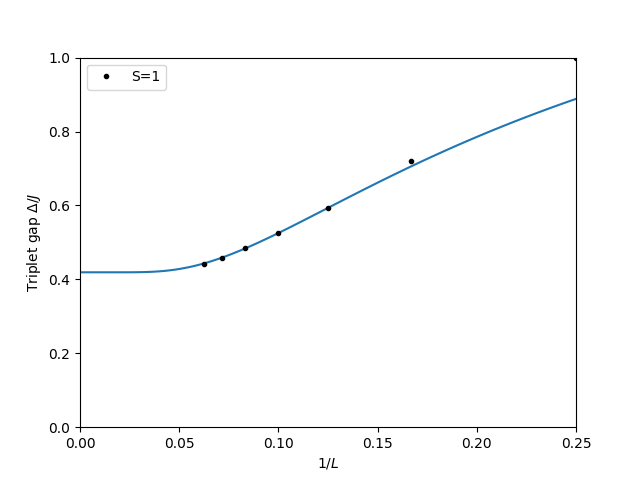
Finite-size extrapolation
The gap at each finite size is:
We plot it against and fit the exponential finite-size correction:
Note that the plot uses as the -axis (so the thermodynamic limit is at ), but the fit function f is written in terms of and called with 1/x. The fw.Parameter objects hold the fit parameters; after fw.fit runs, p[0]() retrieves the fitted value of , p[1]() gives , and p[2]() gives .
We fit only (index [2:] skips and ) to avoid the largest corrections-to-scaling that occur on the smallest chains, where the chain length is not much larger than the lattice spacing:
gapplot = pyalps.DataSet()
gapplot.x = 1./np.sort(lengths)
gapplot.y = [min_energies[(l,1)] - min_energies[(l,0)] for l in np.sort(lengths)]
gapplot.props['xlabel'] = '$1/L$'
gapplot.props['ylabel'] = 'Triplet gap $\Delta/J$'
gapplot.props['label'] = 'S=1'
gapplot.props['line'] = '.'
plt.figure()
pyalps.plot.plot(gapplot)
plt.legend()
plt.xlim(0, 0.25)
plt.ylim(0, 1.0)
# Initial guesses: Delta~0.411 (known Haldane gap), A~1000, xi~6
pars = [fw.Parameter(0.411), fw.Parameter(1000), fw.Parameter(6)]
f = lambda self, x, p: p[0]() + p[1]()*np.exp(-x/p[2]())
fw.fit(None, f, pars, np.array(gapplot.y)[2:], np.sort(lengths)[2:]) # fit L >= 8
x = np.linspace(0.0001, 1./min(lengths), 100)
plt.plot(x, f(None, 1/x, pars))
plt.show()Result
The fitted curve extrapolates to the -intercept at , giving — in good agreement with the best numerical estimates for the Haldane gap (White & Huse, 1993: ). This confirms the Haldane phase and validates the ALPS sparsediag solver.
The plot shows the triplet gap decreasing as grows, with the fitted curve capturing the exponential convergence: