密度矩阵重整化群
什么是 DMRG?
**密度矩阵重整化群(DMRG)由 Steven White 于 1992 年提出,迅速成为求解一维量子格点模型的首选方法。DMRG 在矩阵乘积态(MPS)**的变分空间内寻找基态:
其中每个 是维度至多为 的矩阵。整数 称为键维数(bond dimension),控制近似的精度: 对应乘积态,增大 可以描述更多量子纠缠。
为什么这在一维情况下如此有效?局域哈密顿量的有能隙基态满足面积定律:任意连续格点块的纠缠熵都被一个与块大小无关的常数所限制。这意味着精确基态可以用具有适中固定 的 MPS 很好地近似。对于 海森堡链(无能隙),纠缠熵仅对数增长,因此 – 通常就足够了。
DMRG 通过**扫描(sweeping)**优化矩阵 :从链的一端出发,依次优化每个格点的矩阵(固定其余所有矩阵),然后从另一端扫回,重复直至收敛。每个优化步骤是一个小型本征值问题——远比 ED 所需的完整对角化便宜。DMRG 扫描的计算复杂度为 ( 为局域希尔伯特空间维数),而 ED 为 。对于 、,ED 需要处理约 亿个状态;而 DMRG 在 下完全可行。
为什么选择开放边界条件? DMRG 对开放边界条件(OBC)的链远比对周期边界条件(PBC)更高效。使用 PBC 时,纠缠结构绕链延伸,大约需要两倍的有效键维数才能达到相同精度。因此 OBC 是 DMRG 计算的标准选择。
物理模型
我们研究具有 格点和开放边界条件的 反铁磁海森堡链:
与 链(具有 Haldane 能隙)不同, 链是无能隙的:其低能谱形成 Tomonaga-Luttinger 液体(TLL),具有幂律关联。热力学极限下每个键的精确基态能量由 Bethe 拟设给出:
这是计算量子物理中研究最广泛的基准系统之一,也是学习 DMRG 的理想起点。
运行模拟
完整的设置、运行和分析可以放在一个 Python 脚本中:
import pyalps
import numpy as np
import matplotlib.pyplot as plt
import pyalps.plot
parms = [ {
'LATTICE' : "open chain lattice", # 开放边界条件(更适合 DMRG)
'MODEL' : "spin",
'CONSERVED_QUANTUMNUMBERS' : 'N,Sz', # 利用 N 和 Sz 守恒进行分块对角化
'Sz_total' : 0, # 目标 Sz=0 扇区(基态)
'J' : 1, # 反铁磁交换
'SWEEPS' : 4, # DMRG 扫描次数(左→右→左 = 1 次扫描)
'NUMBER_EIGENVALUES' : 1, # 仅求基态
'L' : 32, # 链长
'MAXSTATES' : 100 # 键维数 m(关键精度参数)
} ]
input_file = pyalps.writeInputFiles('parm_spin_one_half', parms)
res = pyalps.runApplication('dmrg', input_file, writexml=True)参数说明
MAXSTATES是最重要的精度参数,设定最大键维数 。增大 可提高精度,但计算时间以 增长。对于这个 32 格点基准系统, 可使截断误差远低于 。SWEEPS是完整扫描次数(左到右再返回)。这里用 4 次已足以演示收敛;生产计算通常需要 10–20 次,或扫描直至每次能量变化低于阈值。CONSERVED_QUANTUMNUMBERS: 'N,Sz'告诉 DMRG 将总粒子数 和总磁化强度 作为好量子数,对 MPS 矩阵进行分块对角化,使计算更快且数值更稳定。NUMBER_EIGENVALUES: 1仅针对基态。增大此值可让 DMRG 同时针对最低的几个本征态,代价是额外的计算开销。
加载基态可观测量
模拟完成后,loadEigenstateMeasurements 提取 DMRG 代码测量的所有可观测量——包括能量、磁化强度、关联函数以及 ALPS 为最终收敛基态计算的其他物理量:
data = pyalps.loadEigenstateMeasurements(pyalps.getResultFiles(prefix='parm_spin_one_half'))
for s in data[0]:
print(s.props['observable'], ' : ', s.y[0])data[0] 中的每个元素 s 对应一个可观测量。s.props['observable'] 是其名称(如 'Energy'、'Magnetization'),s.y[0] 是其值。对于这个 32 格点开放链,基态能量应接近 。
加载迭代历史
为了了解算法的收敛过程,我们加载每次扫描的测量数据:
itr = pyalps.loadMeasurements(pyalps.getResultFiles(prefix='parm_spin_one_half'),
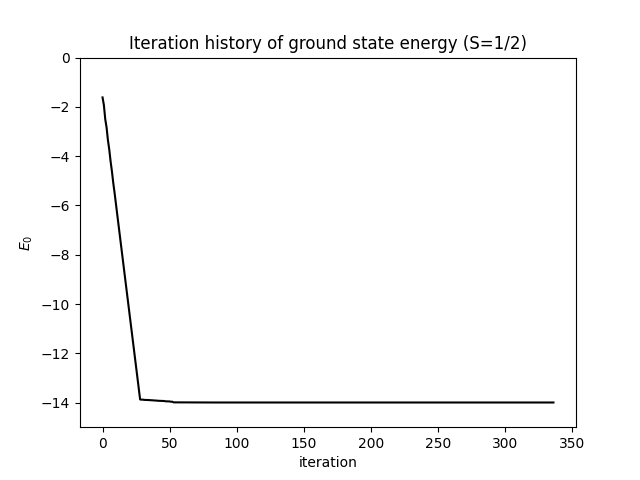
what=['Iteration Energy', 'Iteration Truncation Error'])itr[0][0] 包含每次半扫描记录的能量;itr[0][1] 包含每步的截断误差。截断误差是约化密度矩阵被舍弃本征值之和——即具有 个状态的 MPS 无法表示的精确基态权重。收敛良好的计算应使截断误差低于 ;对于这个系统的 ,典型值在 – 之间。
绘制收敛图
plt.figure()
pyalps.plot.plot(itr[0][0])
plt.title('Iteration history of ground state energy (S=1/2)')
plt.ylim(-15, 0)
plt.ylabel('$E_0$')
plt.xlabel('Iteration')
plt.figure()
pyalps.plot.plot(itr[0][1])
plt.title('Iteration history of truncation error (S=1/2)')
plt.yscale('log')
plt.ylabel('Truncation error')
plt.xlabel('Iteration')
plt.show()结果
能量在前几次扫描内迅速收敛至平台。下图显示了能量随迭代次数的变化——在最后一次扫描之前已趋于稳定:
截断误差在对数坐标下单调衰减,反映了每次扫描后 MPS 近似质量的提升:
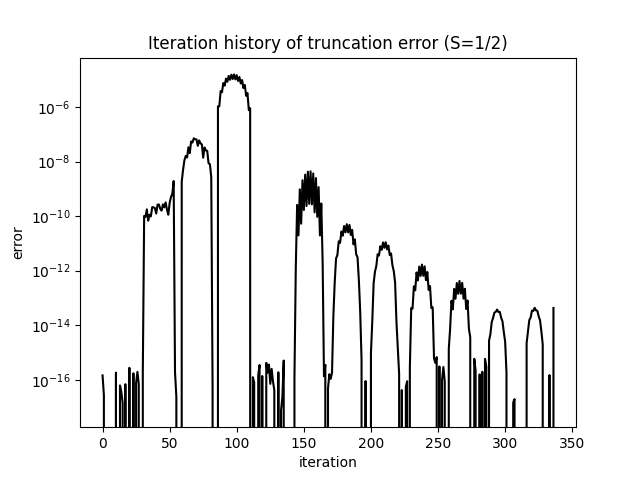
当两条曲线均趋于平坦时,计算已收敛。若要进一步提高精度,可增大 MAXSTATES 并验证能量和截断误差的变化可以忽略不计。